
In our latest expert review, Afiya Andrews discusses the genetics and clinical consequences of dominant-negative growth hormone receptor alterations.
Linear growth is a vital marker of childhood health and well-being, and short stature remains one of the main reasons for referral to paediatric endocrinologists. The approach to assessing children with short stature includes clinical, endocrine and genetic aspects (1). Growth hormone insensitivity (GHI) is identified in approximately 30% of patients with short stature (2).
Patients with short stature may have GHI, defined as normal or raised GH secretion and functional insulin-like growth factor (IGF)-1 deficiency. Laron et al first reported families with classical GHI characterised by severe postnatal short stature as well as typical phenotypic features including mid-facial hypoplasia, protruding forehead, delayed dentition and truncal adiposity (3). The associated biochemical picture is normal or elevated GH, low IGF-1, low IGF binding protein (IGFBP)-3 and undetectable or low GH binding protein (GHBP) levels. This classical presentation, known as Laron syndrome, is uncommon, except in countries with a high rate of consanguinity. Patients may also present with a subtle and non-classical phenotype that can result in diagnostic uncertainty and potentially confused with idiopathic short stature (ISS) (4).
GHI is now recognised to comprise a spectrum of biochemical, phenotypic, and genetic features. The classical genotype associated with GHI is homozygous or compound heterozygous mutations in exons of the gene coding for the extracellular domain of the GH receptor (GHR). Other genetic variants in the GH–IGF-1 axis, including dominant-negative (DN) GHR variants have been shown to cause mild or non-classical GHI (4). Syndromic disorders such as 3M and Noonan syndromes may also present with phenotypic and biochemical features that may overlap with GHI (4, 5). The evolution of next-generation sequencing (NGS) technologies has allowed for the incorporation of genetic testing in the assessment of children with GHI and has expanded our understanding of the genotypes associated with postnatal growth restriction (5).
DN gene mutations create functionally abnormal proteins which impair the normal function of the wild-type protein. To date, 10 DN GHR variants have been identified in children with GHI (6). DN GHR variants are heterozygous and occur in the intracellular or transmembrane domains of the GHR gene (Figure 1). They result in truncated proteins that lack the essential intracellular signalling and internalisation domains. Impaired GHR internalisation contributes to elevated GHBP levels. GHBP is the cleaved extracellular domain of the GHR and binds to circulating GH in a 1:1 ratio. Elevated GHBP is thought to sequester GH and reduce its bioavailability.
Additionally, the GHR exists at the cell surface as a dimer and mutant truncated GHR variants have been shown to form heterodimers with the wild-type GHR (WT) as well as mutant GHR homodimers. The presence of one mutant allele negatively affects the function of the wild-type GHR. These heterozygous GHR variants exert a DN effect on the function of the wild-type GHR with impaired receptor internalisation and downstream signalling through reduced STAT5B phosphorylation.
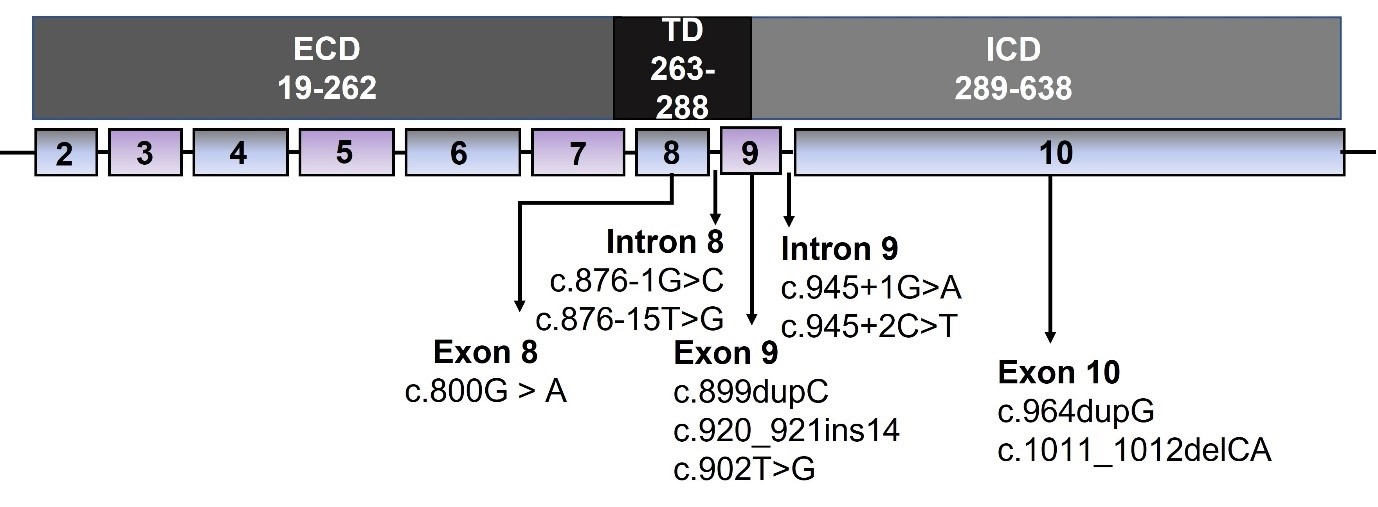
Figure 1. Schematic of the GHR gene structure with the corresponding protein domains and location of published dominant-negative GHR variants. The GHR comprises 638 amino acids encoded by 10 exons and divided into three structural domains.
GHR, growth hormone receptor; ECD, extracellular domain; ICD, intracellular domain; TD, transmembrane domain.
The non-classical GHI phenotype identified in patients with DN GHR variants is characterised by less severe short stature (height standard deviation score [SDS] -2.7 to -4.3) and the absence of the typical Laron syndrome characteristics. Patients have mild-moderate biochemical features of GHI with normal or elevated GH levels, low or normal IGF-1 levels and normal or elevated GHBP levels. This milder phenotype is attributed to the presence of approximately 25% WT GHR homodimers, resulting in delayed diagnosis as patients may seek medical attention at a later stage or because of the challenging clinical workup. Genetic testing is an integral part of the investigation of these patients and should be considered in those with a non-classical GHI phenotype.
The expansion of NGS technologies has widened the access to genetic testing and improved our diagnostic capabilities. Within the UK, National Health Service (NHS) genetic panels are available for patients with short stature. For instance, the R147 panel can be utilised in patients with a height SDS < -3 by the age of at least 2 years. Furthermore, patients who do not fulfil the NHS criteria can also access international research panels such as GRASP (Genetic Research Analysing Short Patients).
Once a diagnosis is confirmed, the mainstay treatment is subcutaneous recombinant human (rh)IGF-1 (mecasermin 0.04–0.12 mg twice daily). Although rhIGF-1 has been shown to improve height velocity, the impact has been varied and less promising than rhGH in GH deficient patients. Further work is therefore required to broaden the treatment options available to patients, such as evaluating GHBP as a potential therapeutic target for short stature (6).
Although the assessment of short stature is common for paediatric endocrinologists, the identification of an underlying aetiology can be challenging. The consideration of genetic pathology such as DN GHR variants in cases of non-classical GHI or idiopathic short stature with instigation of genetic testing is beneficial for early diagnosis and initiation of treatment. It is critical to progress research into the genetic factors impairing growth to reveal additional therapeutic targets for managing short stature.