
In our latest expert review, Catherine Peters gives an update on the incidence, genetic underpinnings, management and outcomes of congenital hypothyroidism.
Newborn screening for Congenital hypothyroidism (CHT) has virtually eliminated the late clinical presentation of this condition, with the associated physical and intellectual sequelae, in the regions of the world where it is available. However, it is important to note that up to 70% of infants are born in areas of the world without access to newborn screening [1].
Screening strategies vary in timing and methodology. In the UK, screening is by heelprick bloodspot Thyroid Stimulating Hormone (TSH) analysis on day 5 of life. Other countries opt for a combined T4/TSH analysis, with a minority designed to detect central CHT in additional to primary hypothyroidism. In all cases, confirmatory thyroid function tests are required for diagnosis. When CHT is diagnosed, thyroid imaging with ultrasound or technetium 99m pertechnate is recommended to aid understanding of the underlying aetiology of the CHT.
After the introduction of screening in the UK, detection of CHT cases significantly increased with a reported incidence of 1:4000 cases [2]. The majority of these cases were due to dysgenesis with profound hypothyroidism. Over time the incidence has increased to nearer 1:2000, in part due to changes of TSH assay, and lowering assay cut points which in turn detects less severe cases. Assay cut points continue to vary between countries and screening laboratories. The incidence of agenesis has remained unchanged and additional cases appear to represent CHT with the thyroid gland in situ. The increased incidence is also linked to ethnicity, population genetics and environmental factors such as iodine deficiency.
A small number of infants will have syndromic CHT and it is important to undertake careful general examination and to document any goitre. In syndromic cases, a clinical genetics opinion and genetic testing should be explored. For other cases with suspected dyshormonogenesis or familial dysgenesis, genetic testing can also be helpful to guide treatment and prognosis [3]. There are over 20 genes implicated in primary CHT. In the UK, the Genomics England R145 panel app for CHT is now available and can be sent after parents have been adequately counselled [4].
As an increasing number of genetic variants are implicated in the development of dyshormonogenesis and dysgenesis, a more complex picture is emerging with an overlap between proteins involved in thyroid hormone synthesis and transcription factors and environmental components such as iodine status [5].
Children with CHT diagnosed by newborn screening would be expected to attain satisfactory neurodevelopmental outcomes. There are a small number of children with this condition who are found to have neurodevelopmental, behavioural and cognitive difficulties. This may be due in part to insufficient in utero thyroxine. Speech and language and communication skills may also be delayed in this group [3].
Levothyroxine treatment should be started as soon as possible. The starting dose of thyroxine depends on initial biochemistry with a recommendation of 10–15 µg/kg per day in severe cases and 5–10 µg/kg per day in milder cases. The aim is to normalise TSH concentrations into the reference range and this may require acceptance of a fT4 slightly above the reference range [3]. Frequent monitoring is required in the first years of life, with thyroxine dose adjustment as needed to maintain euthyroid status [3,7]. Crushed thyroxine tablets or commercial thyroxine solutions are acceptable.
Thyroxine requirements vary over time, with a relatively higher demand in the first few months of life. A small reduction in the efficiency of thyroid hormone synthesis in the neonatal period could result in relative hypothyroidism which improves with time, resulting in “transient” hypothyroidism (fig 1) [6].
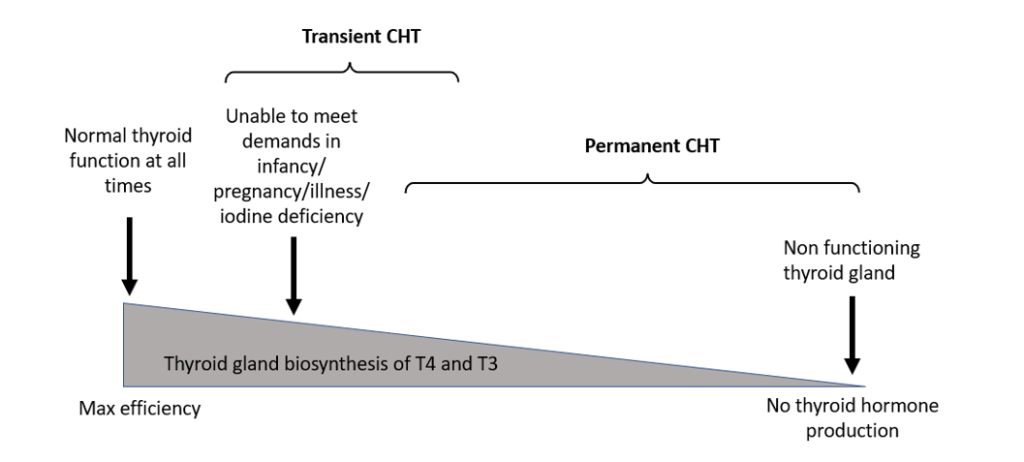
Figure 1. Effect of differences in the efficiency of thyroid hormone biosynthesis
The European and UK guidelines recommend a trial off thyroxine when transient CHT is suspected to avoid unnecessary ongoing treatment [3,7]. In the UK, the CHT clinical referral guidance recommends that “Babies with CHT should undergo a trial off thyroxine at 2 to 3 years of age to establish whether or not they have permanent CHT unless 1 or more of the following conditions are true:
- an isotope or ultrasound scan has demonstrated an absent or ectopic thyroid gland
- there is additional information pointing to a diagnosis of permanent CHT; this might (for example) include the identification of a specific genetic cause of permanent CHT (such as a defect in the gene for thyroid peroxidase or thyroglobulin)
- there is biochemical evidence of a persistent abnormality of thyroid function that is reflected by a rising TSH above 10 mU/L beyond 12 months of age and an associated thyroxine requirement of ≥50 mcg”.
The UK referral guidance also advises feedback of the assessment outcome to the regional endocrine centre to support regional and national audits.
Despite the introduction of newborn screening for CHT over 40 years ago, there are still unanswered questions about the genetics, developmental biology and environmental factors associated with this condition. Clinicians managing children diagnosed with CHT through screening should be aware of the need for close monitoring, optimisation of thyroid doses and re-evaluation during the first 3 years of life.
Discontinuation of thyroxine treatment in children with transient CHT is important to prevent long-term unnecessary treatment. However, some of these cases may be at risk of later hypothyroidism and should be counselled appropriately. This is particularly relevant for female cases who may be at future risk of hypothyroidism during pregnancy [6].