
In our latest expert review, Afiya Andrews outlines the broad phenotypic spectrum of classical and non-classical growth hormone insensitivity disorders in children.
The growth hormone–insulin-like growth factor-1 (GH–IGF-1) axis is essential for normal linear growth. GH binds to the GH receptor (GHR) and activates downstream signalling cascades which result in the transcription of key growth promoting factors including IGF-1 (Figure 1). Genetic defects in the GH–IGF-1 axis have been identified in association with GH insensitivity (GHI). GHI is defined as short stature (height standard deviation score below –2), normal or elevated GH levels and functional IGF-1 deficiency. Defects in the GHR were initially identified with a severe presentation of GHI1. The expansion of next-generation sequencing technologies has since broadened the genetic diagnoses associated with GHI, which is now established to be a clinical and genetic spectrum including genetic defects that are not limited to the GH–IGF-1 axis2.
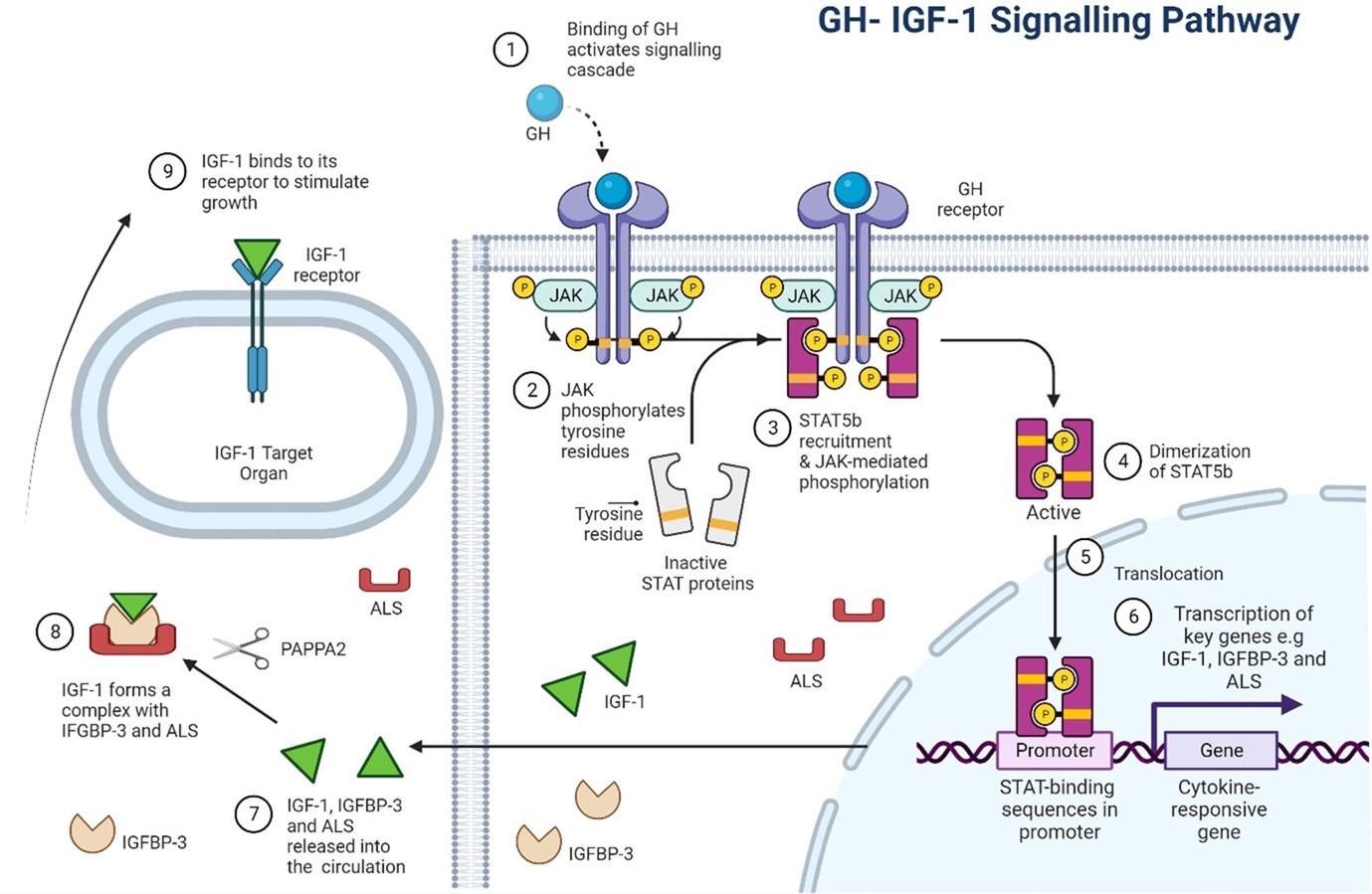
Figure 1: The GH–IGF-1 axis in human growth. GH binds to the GHR resulting in a conformational change to the receptor and activation of the downstream JAK-STAT signalling cascade. Phosphorylated STAT5b is translocated to the nucleus where it regulates the transcription of key growth genes including IGF-1, IGFBP-3 and ALS. IGF-1 exists in the circulation as part of a ternary complex with IGFBP-3 and ALS. PAPPA2 cleaves IGFBP-3 releasing IGF-1 from its complex. IGF-1 then binds to the IGF1R and exerts its downstream growth effects. Defects in this axis have been associated with growth hormone insensitivity. GH, Growth Hormone; IGF-1, Insulin-like Growth Factor 1; IGFBP-3, IGF binding protein 3; ALS, Acid Labile Subunit; PAPPA2, pregnancy associated plasma protein A2. Image created by author with BioRender.com
The classical presentation of GHI, termed Laron syndrome (OMIM #262500), comprises severe short stature, delayed bone age, delayed tooth eruption, delayed puberty and distinctive facial features including frontal bossing, mid-facial hypoplasia and occasionally blue sclera. The biochemical picture is of elevated GH and low levels of IGF-1, GH binding protein (GHBP) and acid labile subunit (ALS). Exogenous administration of recombinant human GH does not restore IGF-1 levels and has no growth-promoting effects. Genetic analysis reveals homozygous or compound heterozygous mutations in the extracellular domain of GHR. Around 100 mutations have been identified to date and there is a high co-relation with parental consanguinity.
In contrast, numerous genetic defects have been identified with an associated milder, non-classical presentation of GHI. These patients have less severe short stature and do not have the classic Laron phenotype. The biochemical profile is milder with normal or elevated GH and GHBP, and normal or low IGF-1 levels. Initially, genetic defects in the GH–IGF-1 axis were identified to account for this presentation but this has since expanded to include additional syndromes not typically associated with a presentation of GHI.
Dominant negative GHR mutations
A dominant negative GHR mutation produces a GHR protein that impairs the action of the wild-type GHR. There have been 10 dominant negative GHR mutations published to date3. These are heterozygous mutations confined to the transmembrane or intracellular domains of the GHR which impair downstream signalling of the GHR. These patients are on the milder end of the GHI spectrum, and their biochemical picture comprises normal or elevated GH and GHBP levels and normal or low IGF-1 levels.
Growth hormone receptor pseudoexons
The GHR 6Ψ4 and 6Ω pseudoexons5 have been identified in patients with GHI. Splicing is the process of removing the intronic non-coding material of DNA. Pseudoexons comprise of intronic sequences that are formed by aberrant splicing. The 6Ψ pseudoexon results in the inclusion of an additional 108 bases between exons 6 and 7 in the extracellular domain of GHR which impairs function of this mutant GHR protein. Patients with this mutation have a phenotypically varied presentation from mild to severe GHI. The 6Ω pseudoexon is formed from the inclusion of 151 bp located between exons 6 and 7, 44 bp downstream of the original 6Ψ pseudoexon. This results in a frameshift and production of a truncated GHR lacking the vital transmembrane and intracellular domains.
STAT5B mutations
Patients with STAT5B mutations (OMIM #245590) have the combined phenotype of GHI and immunodeficiency, related to involvement of the cytokine pathway. Both homozygous and heterozygous mutations have been reported, with the homozygous phenotype being more severe. The biochemical profile comprises normal or elevated GH, normal GHBP and severe deficiencies of IGF-I, IGF binding protein 3 (IGFBP-3), and ALS. Immune dysfunction is varied and includes pulmonary infections and bronchiectasis.
IGFALS mutations
IGF-1 is stabilized in the circulation as part of a ternary complex bound to IGF acid labile subunit (IGFALS) and IGFBP-3 (Figure 1). Homozygous and compound heterozygous mutations in IGFALS (OMIM #601489) have been identified in patients with a mild phenotype of GHI. The biochemical picture is of low IGF-1 and IGFBP-3 which is rapidly cleared due to the IGFALS deficiency. Linear growth is sustained due to normal paracrine IGF-1 production.
PAPP-A2 mutations
Pregnancy-associated plasma protein-A2 (PAPP-A2) cleaves IGF-1 from its ternary complex and releases it into the circulation (Figure 1). Loss of function mutations in PAPP-A2 (OMIM #619485) prevent this cleavage and reduce the amount of free circulating IGF-1. This causes a biochemical picture of elevated IGF-1, IGFBP-3 and IGFALS, however the bioactive form of IGF-1 is low. Patients present at the milder end of the GHI spectrum.
Overlapping syndromes and copy number variants
In addition, defects external to the GH–IGF-1 axis have been identified in patients with GHI. These include 3M, Noonan and Silver–Russell syndromes. Additional overlapping syndromes such as Barth and Bloom syndromes6 and copy number variants have also been identified in patients with GHI7. This highlights the importance of integrating genetic testing as part of the investigative pipeline for these patients.
The phenotypic and genetic presentation of GHI is broad and varied. It includes genetic defects within the GH–IGF-1 axis but has expanded to include other syndromes and genetic diagnoses external to the GH–IGF-1 axis. Awareness of this varied phenotype is essential to guide clinicians to early diagnosis which will aid in the management and prognostication of these patients.