
Find out more about the rare genetic causes of paediatric adrenal insufficiency and how best to treat affected children.
Primary adrenal insufficiency (AI) is a severe endocrine disorder characterised by insufficient glucocorticoid and/or mineralocorticoid and androgen secretion by the adrenal glands, due to impaired adrenal function 1. Genetic causes prevail in the paediatric population. The most frequent genetic cause of PAI is congenital adrenal hyperplasia (CAH), but many other rare genetic diseases, either affecting exclusively the adrenal glands or with systemic involvement, have now been recognized. Underlying mechanisms include defects in signalling, cellular damage, adrenal dysgenesis and syndromic autoimmune disorders, leading to variable severity and presentations.
Defective adrenocorticotropin (ACTH) signalling in the adrenal cortex is the cause of familial glucocorticoid deficiency (FGD), a rare autosomal recessive disorder. The exact prevalence of FGD is unknown. Type 1 FGD is associated with genetic alterations inactivating the gene encoding for the melanocortin receptor 2 (MC2R), while in Type 2 the melanocortin receptor 2 associated protein (MRAP) is affected. A list of FGD variants is available in Table 1. Despite similar mechanisms, phenotypes differ, with Type 1 FGD being associated with early childhood onset and tall stature, whereas Type 2 FGD is characterised by infancy onset and normal height. Shared clinical features include AI, skin hyperpigmentation, recurrent hypoglycaemia, seizures, and failure to thrive.
Alterations of peroxisomal oxidation can lead to adrenal damage due to accumulation of very long chain fatty acids (VLCFA, C≥22). In X-linked adrenoleukodystrophy (X-ALD), caused by variants in the ABCD1 gene encoding for a VLCFA transporter2, the adrenal zona fasciculata and zona reticularis, testicular Leydig cells, and Schwann cells are usually affected and lead to AI, primary hypogonadism and demyelinising disorders of the central and peripheral nervous system. An incidence of 1:21,000 births in boys and 1:16,800 in girls has been reported, although early screening might increase these rates. Prevalence of AI in patients with X-ALD is around 70%, with 57% of X-ALD patients with AI requiring mineralocorticoid replacement2. Nervous system involvement can include cerebral damage and spinal cord injury leading to adrenomyeloneuropathy (AMN), with spastic paraparesis and sensory ataxia.
Allgrove syndrome, also called Triple A syndrome (TAS), is an autosomal recessive disorder characterised by the triad achalasia, PAI, and alacrimia. Less than 50 affected families have been described3. The pathogenesis of TAS is still largely unclear but is mainly caused by variants in the AAAS gene, encoding the protein ALADIN, a nucleoporin scaffold protein of the WD-repeat family, that is mostly expressed in pancreas, adrenal and pituitary gland.
Most patients with TAS present with alacrimia, followed by achalasia (75–85%), including vomiting, weight loss, difficulty to swallow, and coughing, while PAI onset usually occurs within the first 10 years of life. Mineralocorticoid secretion is usually preserved in TAS. Patients may also develop neurological manifestations and autonomic dysfunction.
Autoimmune Addison’s disease and other autoimmune endocrine conditions have been described since the 1950s, configuring autoimmune polyglandular syndromes (APS). APS-1, also known as autoimmune-polyendocrinopathy-candidiasis-ectodermal dystrophy , shows earlier onset and high prevalence in childhood, and is caused by deletions in the autoimmune regulator gene (AIRE)4, which is expressed in thymic medullary epithelial cells and dendritic cells, regulating ectopic expression of tissue-restricted proteins, that allow negative selection of autoreactive thymocytes. The estimated prevalence of APS-1 is 1:100,000 in most countries, while in some regions, such as Finland and Sardinia, it can reach 1:25,000 and 1:19,000, respectively.
“Classical” APS-1 follows recessive inheritance and is characterised by the triad of hypoparathyroidism, PAI, and chronic mucocutaneous candidiasis, with the latter usually being the first presenting conditions and the former the first presenting autoimmunity. Patients can develop more than 20 other minor immune-related conditions and the clinical picture of APS-1 is very variable even among identical genotypes. Also, since “non-classical” presentation due to AIRE heterozygous variants and monogenic autoimmunity may not meet diagnostic criteria for APS-1, it is possible that the prevalence of this disease is underestimated.
PAI due to adrenal dysgenesis and dysplasia can occur in several rare syndromes. The most common cause of congenital adrenal hypoplasia (adrenal hypoplasia congenita, AHC) is the X-linked form caused by germline variant of the nuclear receptor protein DAX-15. Clinical features generally include PAI, hypogonadotropic hypogonadism (HH), and infertility, since the reproductive system is also regulated by DAX-1. PAI is usually observed in the first 2 months of life, presenting as salt-wasting adrenal crises.
Other conditions characterised by adrenal gland dysplasia are intrauterine growth restriction (IUGR) syndromes, with IMAGe, IMAGe-like, and MIRAGE syndromes being the most common.
Conditions associated with isolated mineralocorticoid deficiency are either due to renal or systemic resistance to aldosterone (Type 1 pseudohypoaldosteronism, PHA1), or to altered synthesis of mineralocorticoid due to enzymatic defects (aldosterone synthase deficiency, ASD) and PHA1 is characterised by systemic (type b) or renal (type a) resistance to mineralocorticoid activity leading to neonatal electrolyte imbalances and dehydration, with PHA1b leading to multi-organ salt and water loss and more severe features. Spontaneous clinical improvement can be observed during childhood.
ASD is characterised by variable degrees of hypoaldosteronism (due to complete or partial loss of AS activities, in ASD2 and ASD1, respectively). ASD clinical symptoms usually appear in the first days or weeks of life, with dehydration, hyponatraemia, hyperkalaemia, vomiting, failure to thrive, and sometimes metabolic acidosis.
Children with AI require life-long glucocorticoid and often mineralocorticoid replacement. The suggested starting treatment dose is hydrocortisone (HC) 8 mg/m2 of body surface area, and due to its shorter half-life, HC is preferred over other therapies6. The immediate-release granule formulation of HC, Alkindi®, is now licensed in Europe for glucocorticoid replacement in infancy allowing more accurate dose titration.
Half of the daily dosage should be taken at awakening and one or two additional doses to be taken until 5 pm to mimic the physiological circadian cortisol profile. In children, three or four doses are often required to sustain daily activities and growth. Guidelines suggest that children with AI should be treated with the minimum tolerated dose of GC to avoid the effects of excessive exposure on growth, bone and sexual development 6.
Treatment of acute AI includes fluids and intravenous or intramuscular administration of glucocorticoid. In children, along with rapid bolus of intravenous saline, HC should be administered with an initial bolus of 50–100 mg/m2 followed by 50–100 mg/m2 per day6 . Patients and parents must be trained in glucocorticoid replacement principles during acute illness. The sick child should double the dose of oral HC and if unable to take HC orally, should have intramuscular HC 100 mg administered and seek urgent medical care due to the potential development of life-threatening adrenal crises.
The starting dose of mineralocorticoid replacement (fludrocortisone) in children is usually 0.05–0.10 mg. In conditions of electrolyte imbalance due to real mineralocorticoid deficiency or defective mineralocorticoid action in target tissues, hyponatremia, dehydration and hyperkalemia must be corrected to avoid life-threatening arrhythmias.
AI is a life-threatening condition and prompt diagnosis and treatment are essential in newborn and paediatric patients. Even if genetic causes of PAI are rare, careful clinical observation and biochemical work-up can reduce diagnostic delay, provide adequate counselling to the families, warrant proactive management of concomitant manifestations and improve the outcomes and quality of life of the patients.
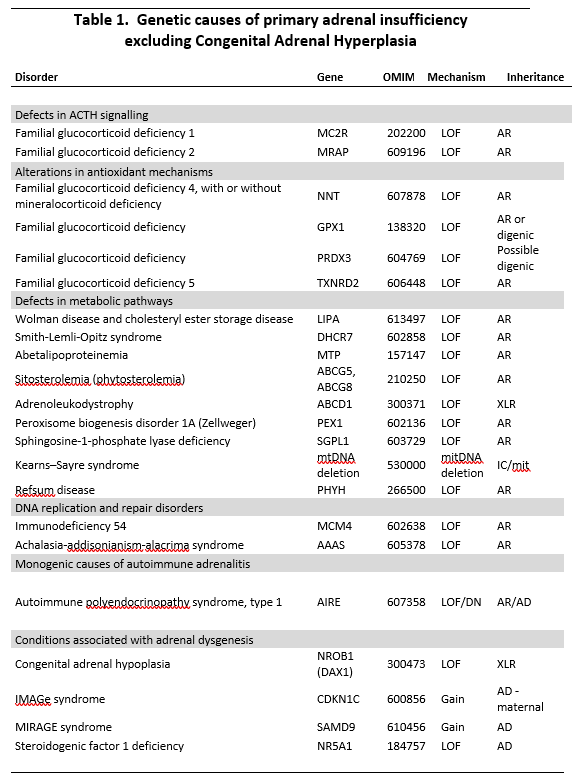
Rare genetic causes of PAI. LOF: loss of function. AR: autosomal recessive. AD: autosomal dominant. DN: dominant negative. XLR: X-Linked recessive
