 The evaluation of children presenting with short stature includes detailed clinical, phenotypic, auxological and biochemical assessments with genetic analyses in selected cases. Advances in molecular technologies and bioinformatic pipelines have broadened the genetic testing available to clinicians and unveiled numerous genetic causes for growth failure. This work has advanced the understanding of the physiology of normal human linear growth, identified new genetic causes of short stature and enhanced patient diagnosis.
The evaluation of children presenting with short stature includes detailed clinical, phenotypic, auxological and biochemical assessments with genetic analyses in selected cases. Advances in molecular technologies and bioinformatic pipelines have broadened the genetic testing available to clinicians and unveiled numerous genetic causes for growth failure. This work has advanced the understanding of the physiology of normal human linear growth, identified new genetic causes of short stature and enhanced patient diagnosis.
Approximately 80% of children referred with short stature do not obtain a specific diagnosis despite detailed evaluation and many children with undiagnosed short stature have normal growth hormone (GH) secretion. The finding of a low serum IGF-I concentration in association with sufficient GH levels, particularly when there is severe short stature, is suggestive of growth hormone insensitivity (GHI) (1). Evidence of GHI is reported in approximately 30% of children referred for investigation of short stature.
Growth hormone insensitivity (GHI) in children is characterised by short stature, functional IGF-I deficiency and normal or elevated serum GH concentrations. GHI encompasses a spectrum of defects of GH action presenting in childhood with growth failure. In its severe form, GHI is associated with extreme short stature, dysmorphic features and metabolic abnormalities (2). In recent years, the clinical, biochemical and genetic characteristics of GHI and other overlapping short stature syndromes have rapidly expanded. This can be attributed to advancing genetic techniques and a greater awareness of this group of disorders.
Genetic defects of the GH-IGF-I axis are an established cause of GHI; however, their exact prevalence is not well established. “Laron syndrome” or “classical” GHI due to homozygous or compound heterozygous defects of the GH receptor gene (GHR) presents at the extreme end of the spectrum with marked postnatal growth failure and IGF-I deficiency due to severe GH resistance. To date, more than 90 homozygous, compound heterozygous, missense, nonsense, and splice site GHR mutations have been identified with significant phenotypic and biochemical variability (3).
Genetic defects have also been discovered in key downstream GH-IGF-I axis genes such as STAT5B, IGFI, IGF2, IGFALS and PAPPA2 and we can now conceptualize a spectrum of GHI presentations in patients from very mild to very severe. Each known defect in the GH pathway has a distinct clinical, biochemical, metabolic and/or genetic signature. Other molecular defects impacting GH signalling and causing GHI phenotypes include STAT3, IKBKB, IL2RG, PIL3R1 and FGF21 mutations (4).
Our recent published series of patients presenting with short stature and apparent GHI, indicated that genetic variants in the GH-IGF-I axis comprised the most common cause, accounting for 56% of patients in whom a diagnosis was made (1). This was not unexpected, given the biochemical phenotype and high incidence of consanguinity (34%) in our cohort.
Interestingly, we also identified a wide range of additional genetic diagnoses and 44% had “overlapping” short stature syndromes/disorders (1). This included single diagnoses in individuals of nine disorders not previously linked to GHI. Patients with diagnoses external to the GH-IGF-I axis were more likely to be born small for gestational age as many of these disorders, such as 3M and Silver-Russell syndromes, cause both pre- and post-natal growth restriction.
GHI comprises a spectrum of clinical entities caused by genetic defects in and external to the GH-IGF-I axis. Clinical diagnosis can be challenging as the predominant, consistent feature of many of these conditions is short stature. The associated dysmorphic features can frequently be subtle, non-specific and overlap with other disorders. Detailed clinical and genetic assessment may identify a diagnosis and inform clinical management (5). The identification of an underlying genetic defect can enable access to treatments, genetic counselling, early detection of likely comorbidities and will inform prognosis.
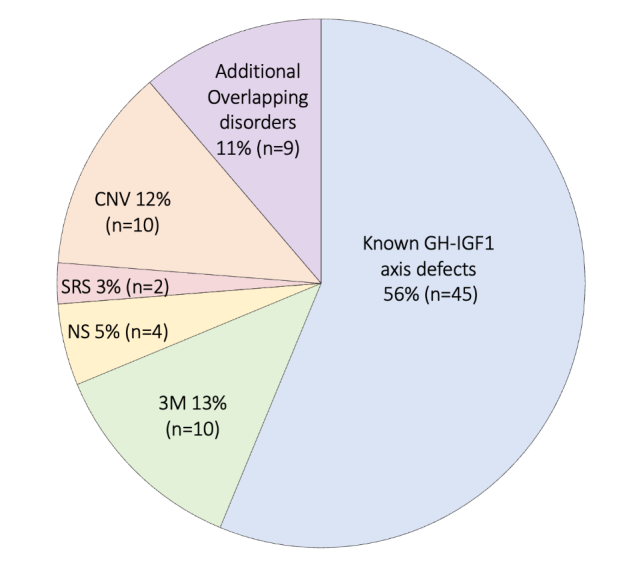
Figure 1. The range of genetic diagnoses identified in a cohort of 149 patients with short stature and suspected growth hormone insensitivity
Genetic diagnoses were identified in a total of 80/149 individuals. Of the 80 with a genetic diagnosis: 45 had growth hormone-insulin-like growth factor-I (GH-IGF-I) axis gene variants (GHR n=40, IGFALS n=4 and IGFIR n=1), 10 had 3M syndrome genetic variants (OBSL1 n=7, CUL7 n=2 and CCDC8 n=1) and four had Noonan syndrome (NS) genetic variants (PTPN11 n=2, SOS1 n=1 and SOS2 n=1). Two patients were diagnosed with Silver-Russell syndrome (SRS) (Loss of methylation on chromosome 11p15, uniparental disomy for chromosome 7). Class 3-5 copy number variations (CNVs) were identified in 10 patients (Class 4 1q21 deletion n=2, Class 5 12q14 deletion n=1, Class 3 5q12 deletion n=1, Class 4 Xq26 duplication n=1, duplication of Chromosome 10 n=1, Class 3 7q21 and Class 4 7q31 deletion n=1), Class 3 7q21 duplication and Xp22 duplication n=1, Class 3 7q36 duplication n=1, Class 3 3p22 deletion and 15q13 duplication n=1). Additional overlapping disorders were diagnosed in nine individuals (Barth syndrome, Autoimmune lymphoproliferative syndrome, Microcephalic osteodysplastic primordial dwarfism Type II, Achondroplasia, Glycogen storage disease Type IXb, Lysinuric protein intolerance, Multiminicore disease, MACS syndrome and Bloom syndrome).