INCREASING HEIGHT VELOCITY
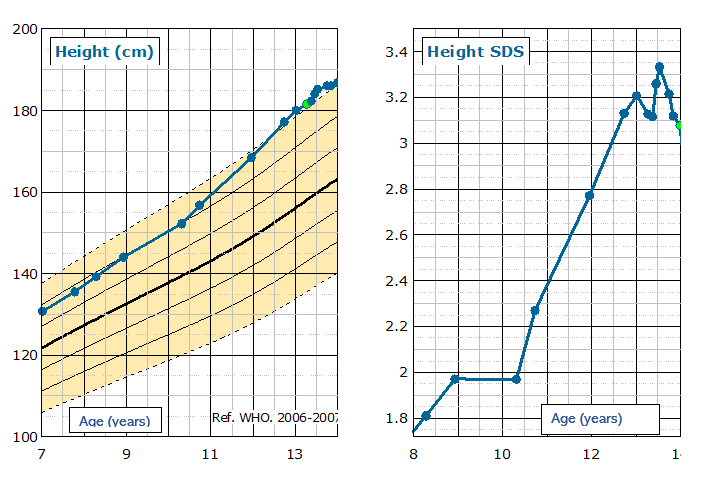
The patient continued to grow quickly and at 11.1 years auxology was:
- Height 168.4 cm. +2.8 SDS.
- Height velocity 9.5 cm/year

However, IGF-1 remained in the normal range:
| Month | IGF-1 (NR 18.6–65.8 nmol/L) |
| March 2022 | 55.5 nmol/L |
| April 2022 | 60.2 nmol/L |

- Height measurements
- IGF-1 during treatment
- Glycated haemogobin (HbA1c)
- Side effects
- MRI scans to monitor size of adenoma.
PATIENT HISTORY DURING LANREOTIDE TREATMENT
- Patient reviewed by paediatric endocrine nurse specialists once a month for height measurements, blood tests and lanreotide injection.
- Patient suffered abdominal cramps and diarrhoea when he started treatment and after dose increases. Dose was occasionally reduced to help with side effects and loperamide also used to alleviate symptoms. Side effects settled over time.
- Over a period of 10 months the monthly dose was increased from 60 mg to 90 mg and then 120 mg.
- Size of adenoma reduced from 14 mm x 18 mm x 15 mm to 10 mm x 14 mm x 6 mm.
- IGF-1 remained elevated despite dose increases, however his growth stopped, and his epiphyses looked mature on X-ray.
Table 3: IGF-1 and growth response to treatment with lanreotide
| Months after the start of treatment | IGF-1 (NR 18.6–65.8 nmol/L) | DOSE OF LANREOTIDE (mg) | HEIGHT (cm) | HEIGHT SDS |
| Baseline | X | 60 | X | X |
| 1 | 70.2 | 40 | 181.5 | 3.13 |
| 2 | 67.0 | 60 | 182.3 | 3.12 |
| 3 | 61.2 | 60 | 184.0 | 3.26 |
| 4 | 69.3 | 60 | 185.2 | 3.33 |
| 5 | 73.3 | 90 | X | X |
| 6 | 72.4 | 90 | X | X |
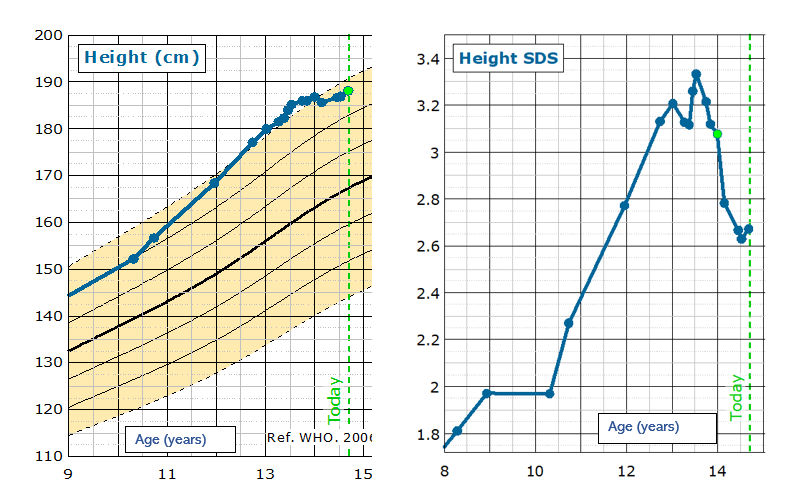
| 7 | 75.4 | 120 | 186.0 | 3.21 |
| 8 | 73.5 | 120 | 186.0 | 3.12 |
| JANUARY 2024 | 78.6 | 120 | 186 | 3.08 |
- Height measurements
- IGF-1 during treatment
- LFTs
- Continue monitoring pituitary hormones as the adenoma could cause other pituitary dysfunctions
- MRI monitoring of adenoma.
PATIENT TODAY
- Patient and family trained on administration of pegvisomant by a paediatric endocrine nurse specialist, and he now self-administers all injections.
- Currently patient has had no side effects of pegvisomant demonstrated in biochemistry or physical examination.
- IGF-1 levels demonstrated good initial response to pegvisomant.
- When IGF-1 level was noted to have increased, the dose of pegvisomant was increased to 15 mg/day as per adult guidance (Table 4)
Table 4: IGF-1 response to the introduction of pegvisomant
| Months after the start of medical treatment | IGF-1 (NR 18.6–65.8 nmol/L) | TREATMENT |
| 12 | 89.2 | lanreotide 120 mg/day |
| 13 | 85.8 | lanreotide 120 mg/day |
| 14 | 74.8 | pegvisomant 10 mg/day |
| 15 | 62.4 | pegvisomant 10 mg/day |
| 16 | 71.4 | pegvisomant 15 mg/day |
| 17 | 61.2 | pegivsomant 15 mg/day |
- Patient’s height is static and growth almost complete.
- Patient additionally developed secondary adrenal insufficiency (peak cortisol of 348 nmol/L on synacthen testing) and secondary hypothyroidism. He was started on hydrocortisone and levothyroxine which continues to be monitored by the endocrinologist.
- He continues to be monitored with height, weight, bloods and blood pressure checked by paediatric endocrine nurse specialists on a monthly basis.
- His dose of pegvisomant may continue to be increased if IGF-1 levels rise.
